Reactive Molecular Dynamics
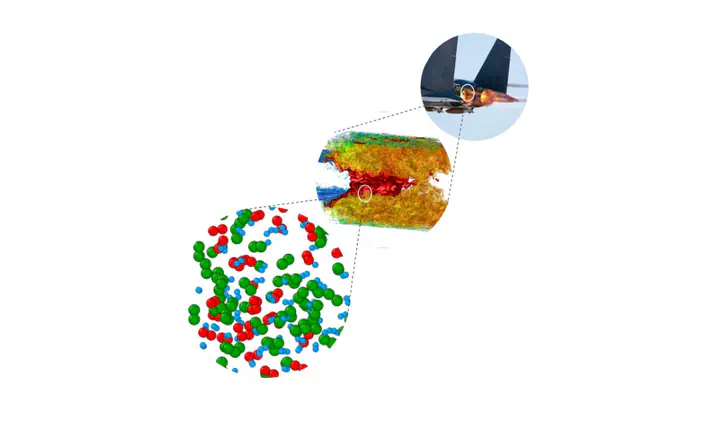
Chemical kinetic (CK) is a vital part of combustion system modeling. It provides reaction mechanisms and rate constants extracted from theoretical approaches. However, CK models are only valid in a specific range of temperatures and pressures and eventually fail at extreme conditions. Reactive molecular dynamics (MD) can incorporate the complexities at extreme conditions and provide the entire mechanism as well as the parameterization required for the combustion systems. MD is often performed at two levels: reaxff-based MD (RMD) and ab-initio MD (DFT-MD). RMD has lower compute costs, but it is less accurate than DFT-MD. We perform CK modeling of the hydrogen-oxygen system using RMD and DFT-MD to extract the complete reaction mechanism and the statistically computed rate constants from atomistic simulations. Arrhenius parameters are then fitted for each elementary reaction using the computed kinetic rates at different temperatures, and a detailed CK model is generated for hydrogen oxidation. The parametrization obtained from both the MD approaches are evaluated by computing the flame speed of a one-dimensional premixed in continuum scale simulations and compared against standard results, showing good agreement.